
Research
Proteins are the molecular basis of life, and yet, we lack reliable means to predict what they do, how they do it, where and when. Our research focuses on the development and application of machine learning algorithms for elucidating the relationship between the sequence, structure, function and evolution of proteins. We then apply these tools to computationally design new proteins! See below some recent works:
Geometric deep learning for functional site annotation
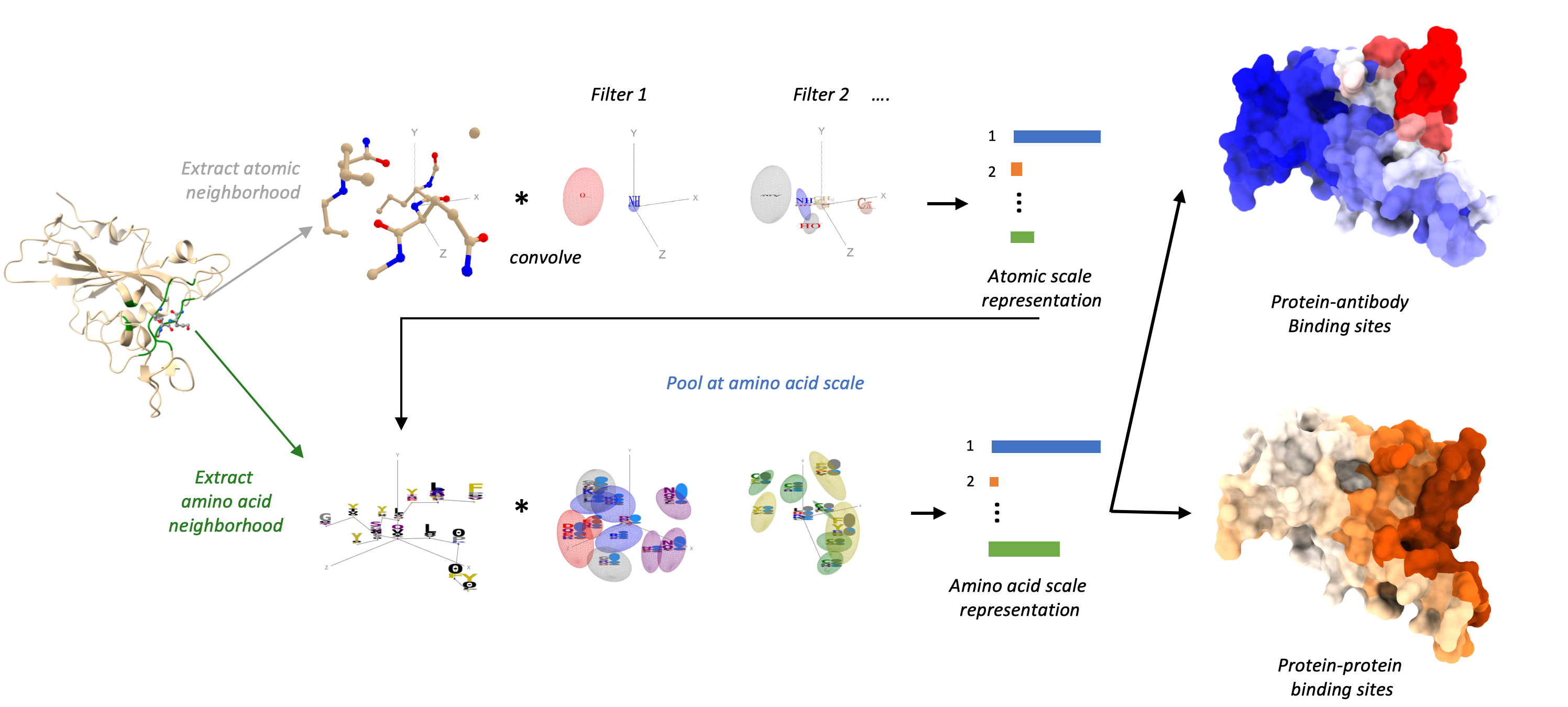
Deep learning-based protein structure prediction algorithms such as AlphaFold are accelerating biology and drug discovery research by providing access to reliable protein structure models at scale. However, human experts often struggle to gain insights into the function of a protein by visual inspection of its structure. We recently developed ScanNet (Spatio-Chemical Arrangement of Neighbors Network), an interpretable, multi-scale geometric deep learning architecture tailored for protein structures. ScanNet builds representations of atoms and amino acids based on the spatio-chemical arrangement of their neighbors. We trained ScanNet for detecting various classes of functional sites, and demonstrated superior performance compared to traditional machine learning or homology methods. The network learns structural motifs of interest directly from raw data; we found that it detects simple, generic structural motifs such as hydrogen bonds or solvent exposed hydrophobic side-chains, whereas others recognize complex, task-specific motifs. See the original article and the research briefing for more information. More broadly, we aim to leverage geometric deep learning to decipher the link between the structure and function of proteins.
Generative models for peptide binder design
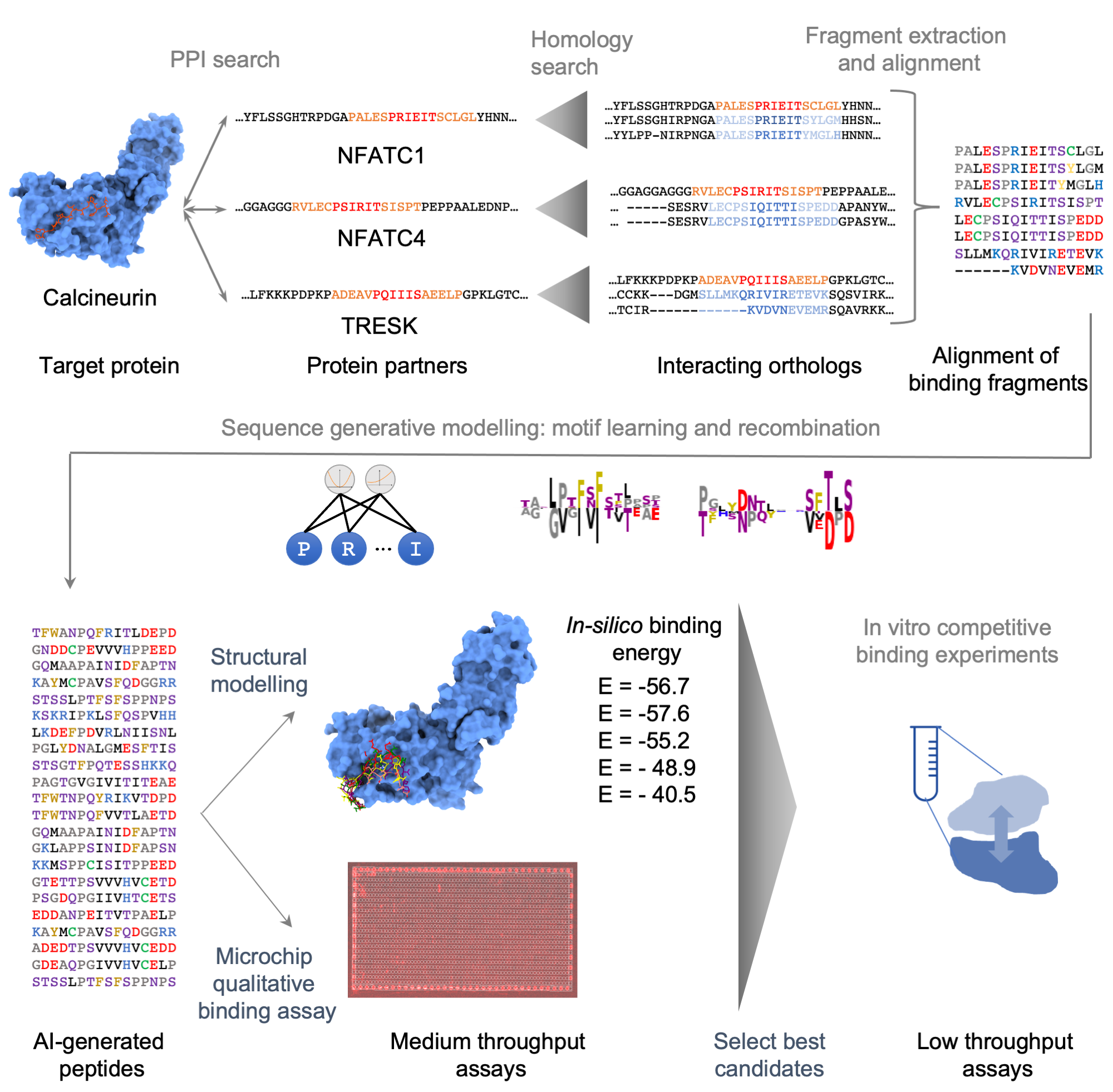
Protein-protein interactions (PPIs) play a critical role in countless molecular pathways. As such, interfering with PPIs is a major therapeutic strategy, with numerous examples such as the celebrated PD1/PDL1 immune checkpoint inhibitors in immunotherapy. Peptides binders are short proteins (L<20 amino acids) that bind the interaction site of a protein and prevents the native PPI from occurring. Designing such peptides is however, a major challenge. We developed an integrative peptide binder design discovery protocol based on a sequence generative model. The generative model (a compositional Restricted Boltzmann Machine) is trained on known and putative protein binders of the target protein identified from previous experiments and homology search on large sequence databases. The model “reverse-engineers” the key sequence motifs underlying binding, and generates novel sequences by recombination of these motifs. We tested our protocol on Calcineurin, an important activator of the immune system, and discovered in a single screening round multiple novel peptides capable of interfering with Calcineurin PPIs. See the research article here . Our aim is to develop new computational methodologies for peptide design based on generative models and geometric deep learnings.